5.1 概要
医薬品・医療機器規制の概要
医薬品・医療機器制度を構成する中核的な法令は、「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」(薬機法)である。医薬品や医療機器は人体に直接作用するため、生命や身体に対する危害を及ぼす可能性が高いことから、薬機法による規制が必要とされている。規制の程度は、対象となる医薬品・医療機器の分類によって異なる。
| 規制区分 | 主な分類や具体的な品目例 |
|---|---|
| 医薬品 | 医療用医薬品 |
| 市販薬(要指導医薬品、一般用医薬品(第1類〜第3類医薬品)) | |
| 体外診断用医薬品(例:血液学的検査薬等) | |
| 医薬部外品 | うがい薬、殺虫剤、染毛剤、栄養ドリンク等 |
| 化粧品 | 一般的な化粧品、シャンプー、スキンケア用品等 |
| 医療機器 | ペースメーカー、人工関節、超音波画像診断装置、メス |
| 再生医療等製品 | 細胞加工製品(例:心筋の細胞シート等) |
| 遺伝子治療用製品(例:欠損した遺伝子を人の体内に投与するもの) |
医薬品や医療機器は、研究開発から販売後の影響に至るまで、各段階に応じた規制を受ける。具体的にどのような内容の規制を受けるのかは、規制対象の分類に応じて異なる。
| 1 | 開発・治験 | 医薬品等の品質、有効性、安全性を確保するための臨床試験の方法やデータの集め方等を規制 |
| 2 | 承認審査 | 医薬品医療機器総合機構(PMDA)が品質、有効性及び安全性を審査 薬事審議会からの答申を受け、厚生労働大臣が承認 |
| 3 | 製造 | 品質確保の観点から、製造業、製造販売業を規制 |
| 4 | 販売規制 | 医薬品等の流通経路(薬局、店舗販売業等)を規制 医薬品等の表示(ラベルや外箱、添付文書)を規制 |
| 5 | 市販後安全対策 | 副作用等の情報収集 副作用の拡大を防ぐための安全対策(添付文書の改訂等)の実施 |
| 6 | 監視指導 | 無承認・無許可医薬品の監視指導 不良医薬品等の取締り |
| 7 | 副作用被害の救済 | 医薬品副作用被害救済制度等による給付 ※独立行政法人医薬品医療機器総合機構法で規定 |
現在の医薬品・医療機器規制の骨格は、品質・有効性・安全性を確保するための規制を定めた1960年の旧薬事法の全面改正によって形作られた。その後2013年に大幅な改正が行われ、法律の名称が現在のものに変更された。この改正では、医療機器の特性を踏まえた規制の創設や、再生医療等製品の承認制度の整備などが行われた[1]。近年では再生医療等製品の製造販売に関する規制や、緊急時の薬事承認、オンライン資格確認を基盤とした電子処方箋の仕組み等の規定が追加されている[2]。
医薬品・医療機器の流通と価格
上記の医薬品・医療機器規制の全体の中で、ここでは主に流通(製造・販売)と価格決定について説明を行う。医薬品・医療機器を国内で流通させるための規制は、品目と事業者の二種類に分類できる。これらを通過することではじめて、医薬品・医療機器を市場に流通させることが認められる。
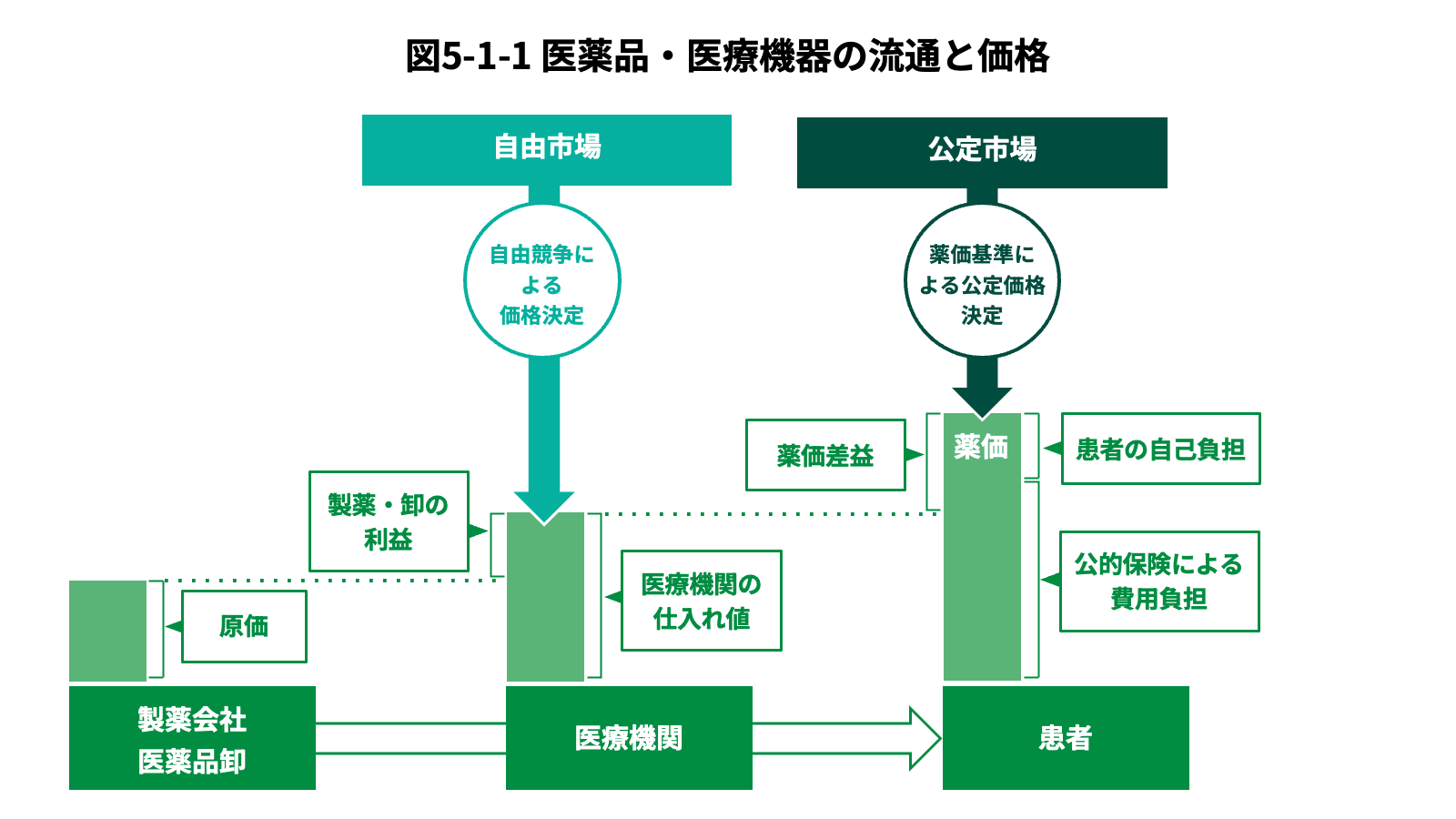
市場に流通した医薬品・医療機器の価格は、公定価格によって決まるものと自由市場によって決まるものがある。この公定価格とは、公的医療保険制度に基づく保険診療において提供される医薬品や医療機器の価格であり、政府が価格を決定する。Section 1.2で確認した通り、日本国内の医療の大部分が保険診療であるため、医療用の医薬品・医療機器は政府による価格決定の影響を強く受ける。他方で、医療用ではない市販薬や家庭用の医療機器は、自由市場で価格が決定される。
実際の保険診療では、自由市場による価格決定と公定価格による価格決定の組み合わせとなっている。特に医薬品についてはこの仕組みが度々制度改革における大きな論点となる。具体的には、病院や診療所、薬局などの医療機関が患者へ提供する医療サービスは公定価格によって支払われる一方で、医療機関が医薬品の卸業者から購入する際には自由市場によって価格が決定される。医薬品の卸業者が製薬企業から購入する場合も自由市場である。一般に、自由市場による取引の方が公定価格よりも安価で行われることが多い。そのため医療機関は、仕入価格と公定価格の差による利益(薬価差益)を得ることができる。そうしたこともあり政府は、薬価差益が大きい場合には公定価格の引き下げ余地があるとして、薬価改定時に薬価を引き下げることがある。一方で近年は、原材料費や人件費、配送費などの高騰により、仕入価格が公定価格を上回る状況(逆ザヤ)も一定程度生じており、医療機関の経営を圧迫する要因となっている。
薬事承認から保険適用までの流れ
次に、薬事承認から保険適用までの流れの概要を説明する。薬事承認とは、医薬品・医療機器の有効性や安全性を担保するために規制当局によって承認を受ける薬機法上のプロセスである。また、保険適用とは、薬事承認ののちに公定価格を決定し、保険適用範囲に含める医療保険制度上のプロセスである。なお、各ステップでの詳細については、Section 5.2以降の各セクションで医薬品・医療機器の分類ごとに詳述する。
- 日本では、医薬品や医療機器の製造販売にあたり、厚生労働大臣に承認申請をして認められなければならない。この際、承認するのは厚生労働大臣だが、有効性や安全性などの審査はPMDA(独立行政法人医薬品医療機器総合機構)が担う。ここで承認を得た医薬品・医療機器は使用が可能となる。
- ただし、承認だけでは公的医療保険は適用されない。Section 1.2で述べたように、日本は保険適用医療が医療給付の大半を占めているため、実際に医療機関等で利用されるためには、医薬品・医療機器の公定価格が決定され、保険適用範囲に含まれることが重要であることが多い。
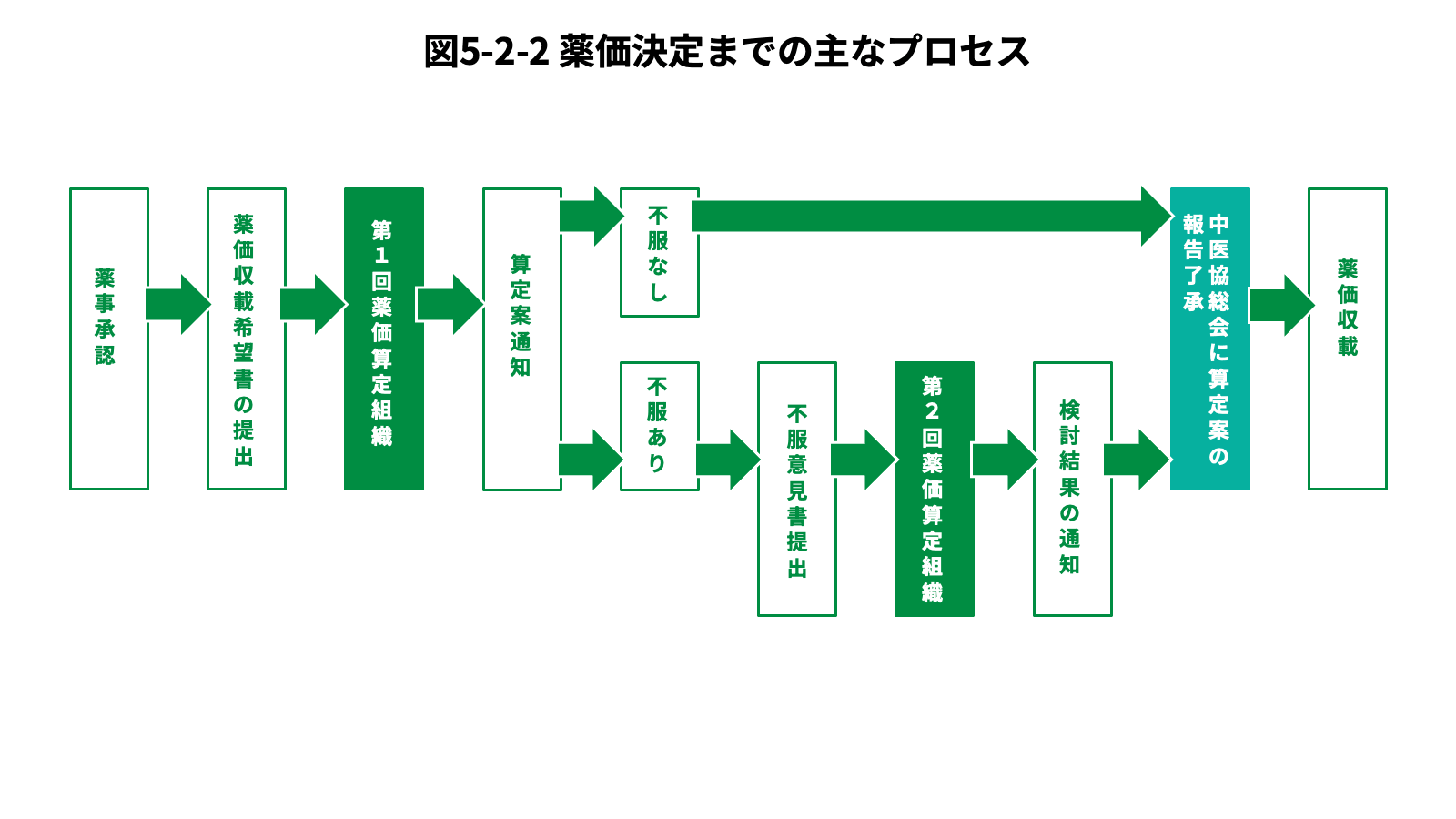
- 医薬品の場合、製薬会社は承認を得た後、保険適用のための申請を厚生労働大臣宛てに提出し、大臣の諮問機関である中央社会保険医療協議会(中医協)が医薬品の公定価格である薬価を査定する。ここで決定した薬価は、公定価格である薬価をまとめたリストである「薬価基準」に追加され、保険適用の対象となる[3]。効能が類似している既存薬の薬価や、企業が算出した原価などを基に薬価が決まり、保険適用される。収載時期は承認後原則60日以内、遅くとも90日以内とされている。なお、医療用医薬品については原則保険適用される[4]。
- 医療機器の場合、医療機器の保険上の評価区分に応じて保険適用の手続きが異なる。その概略はSection 5.5にて記載するが、評価区分が複雑であるため、厚生労働省はPMDAによる「SaMD一元的相談窓口(医療機器プログラム総合相談)」[5]を活用することで評価区分を確認することを推奨している[6]。
医薬品・医療機器の市場規模[7]
世界市場に占める日本の市場規模は、医薬品において4.2%程度[8]、医療機器において5.0%程度[9]だと言われている。
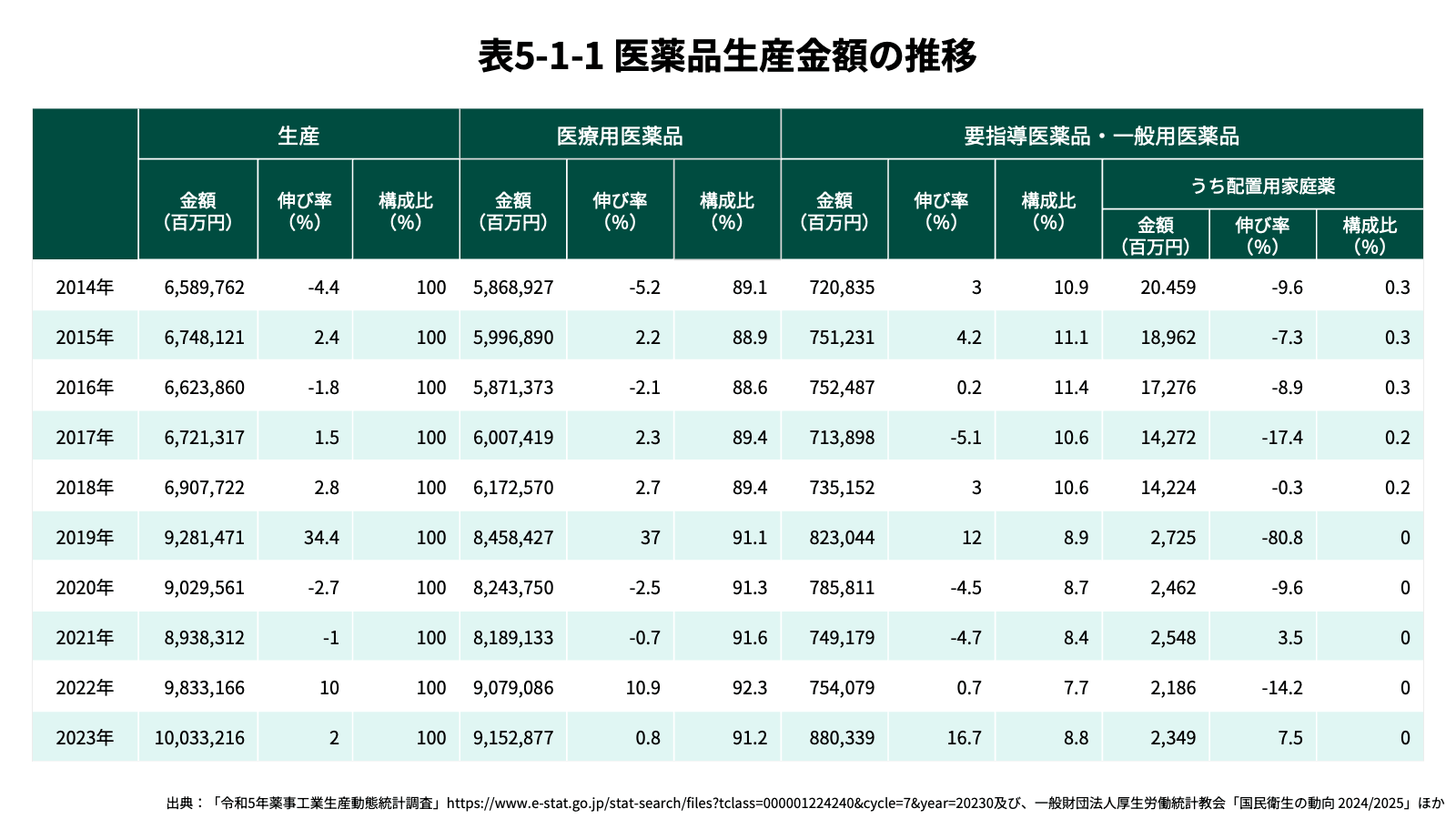
日本の医療用医薬品の市場規模は拡大傾向にあり、2023年時点での国内生産金額は10兆円程度に至っており[10]、外国からの輸入金額の約3.8兆円を合わせると、合計で14兆円弱である。要指導医薬品・一般用医薬品の生産規模は、約8,800億円と増加傾向にあり、医薬品の国内生産金額全体のおよそ10%を占める。
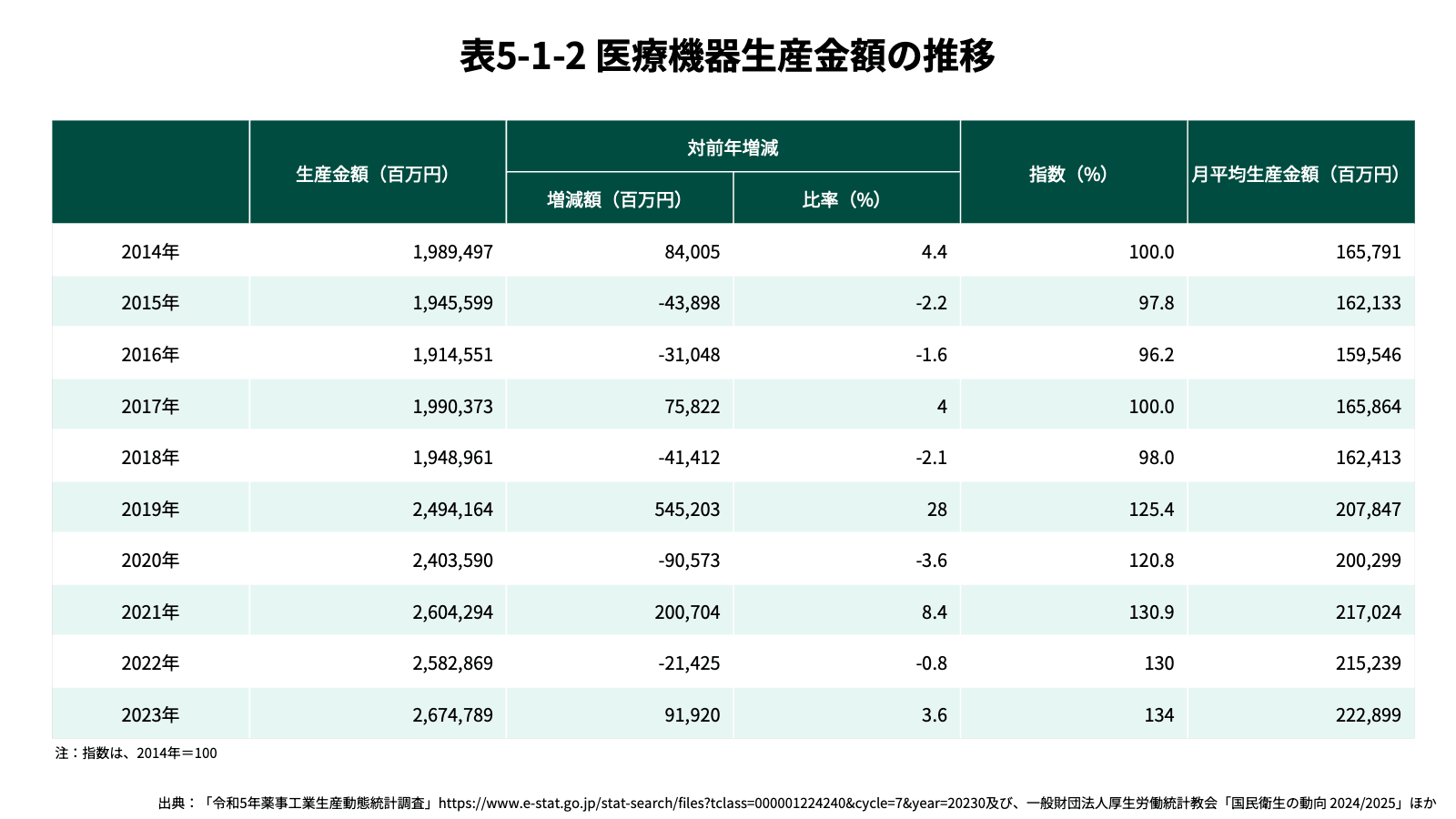
国内医療機器の市場規模も拡大を続けており、2023年時点での国内生産金額は約2.7兆円(前年比3.6%増)であり、輸入金額は約3.3兆円(前年比13.8%増)で、合わせて6兆円弱である。一方で、医療機器市場は医薬品と比較して輸入に依存した構造になっている。
医薬品は、大きく医療用医薬品と一般用医薬品に分けられる。医療用医薬品は更に、先発医薬品と後発医薬品(ジェネリック医薬品)に分けられる。それぞれの定義を以下に示す。
| 医療用医薬品:医師または歯科医師によって使用され、またはこれらの者の処方箋もしくは指示によって使用されることを目的として供給される医薬品。 | 先発医薬品:新しい効能や効果を有し、臨床試験などにより有効性や安全性が確認され、承認された医薬品[11]。 |
| 後発医薬品(ジェネリック医薬品):先発医薬品の特許が切れた後に、先発医薬品と成分や規格が同一で、治療学的に同等であるとして承認される医薬品。 | |
|
要指導医薬品:販売・購入の際に処方せんは不要であるが、薬剤師が対面で情報提供や指導などを行うことが義務付けられ、インターネット販売ができない医薬品。 ※詳細は、Section 5.4―一般用医薬品にて記載。 |
|
| 一般用医薬品:上記以外の医薬品。 | ― |
Section 5.2以下では、各医薬品カテゴリと医療機器について、薬事承認とそれに続く保険適用という流れに沿って、制度の概要および論点を詳述する。
- [1] 厚生労働省「薬事法等の一部を改正する法律の概要(平成25年法律第84号)」https://www.mhlw.go.jp/file/06-Seisakujouhou-11120000-Iyakushokuhinkyoku/0000066816.pdf
- [2] 米村滋人「医事法講義(第2版)」、一般財団法人厚生労働統計教会「国民衛生の動向 2024/2025」ほか
- [3] 薬価基準については、厚生労働省「薬価基準収載品目リスト及び後発医薬品に関する情報について(令和7年3月31日まで)」https://www.mhlw.go.jp/topics/2024/04/tp20240401-01.htmlにて確認できる。毎年最新のウェブページに更新されるため、注意されたい。
- [4] 一方で、直ちに生命にかかわる病気を治療するものではなく、生活の質を改善する目的で使用される一部のものは「生活改善薬」と呼ばれ、通常は保険の適用外となる。
- [5] PMDA「SaMD一元的相談窓口(医療機器プログラム総合相談)」https://www.pmda.go.jp/review-services/f2f-pre/strategies/0011.html
- [6] 厚生労働省「令和6年度医療機器・体外診断用医薬品の保険適用に関するガイドブック」https://www.mhlw.go.jp/content/10800000/001073851.pdf
- [7] 特段の記載がない限り、本項で用いている統計データは「令和5年薬事工業生産動態統計調査」https://www.e-stat.go.jp/stat-search/files?tclass=000001224240&cycle=7&year=20230及び、一般財団法人厚生労働統計教会「国民衛生の動向 2024/2025」ほか を参照している。
- [8] IQVIA INSTITUTE, Global Use of Medicines OUTLOOK through 2029 をもとに算出。
- [9] 経済産業省医療機器産業ビジョン研究会「医療機器産業ビジョン 2024」
- [10] 2023年の数値が大きく伸びているように見えるが、これは統計調査の手法変更に伴い回収率が大幅に向上したことなどの影響が大きい。
- [11] 厚生労働省「薬価基準収載品目リスト及び後発医薬品に関する情報について(令和7年8月14日適用)」https://www.mhlw.go.jp/topics/2025/04/tp20250401-01.html
5.2 先発医薬品
先発医薬品の定義
先発医薬品(新医薬品)とは、医薬品医療機器法第十四条の四に定められる既に承認を与えられている医薬品と有効成分、分量、用法、用量、効能、効果等が明らかに異なる医薬品である。主に、新有効成分含有医薬品、新医療用配合剤、新投与経路医薬品、新効能医薬品、新剤型医薬品、新用量医薬品などを指す[12]。
医療用医薬品の承認
製薬会社が新医薬品を製造、配送、販売するためには、以下のステップを踏むことになる。まず、PMDAから販売承認を得る必要がある。PMDAは、臨床試験を通じて得られた新医薬品の効果と、安全性への評価情報に基づいて承認を行う。臨床試験データ及び提出される書類の書式は、厚生労働省が定める品質、医薬品の安全性に関する非臨床試験の実施の基準(GLP: Good Laboratory Practice)、医薬品の臨床試験の実施の基準(GCP: Good Clinical Practice)に従う必要がある。なお、PMDAによる承認審査は、申請受付から承認までの総審査期間として、標準的なプロセスにおいては12ヶ月という目標を掲げている[13]。そしてPMDAの承認が得られると、薬事審議会において新医薬品の承認可否が検討され、その答申に従って厚生労働大臣が承認可否を決定するという流れである。
また近年は、通常の承認プロセスに加えて、画期的な新薬の開発を促進できるよう、優先的に審査・承認する制度も導入している。その1つが、2014年に導入された「先駆的医薬品指定制度」である。これは、患者に世界で最先端の治療薬を最も早く提供することを目指し、日本で開発され、かつ日本で初めて登録申請される製品への審査・承認を優先的かつ短期間で行う仕組みである。この対象となるためには、新医薬品が1. 治療薬の画期性、2. 対象疾患の重篤性、3. 対象疾患に係る極めて高い有効性、4. 世界に先駆けて日本で早期開発・申請する意思・体制[14]という4つの条件を満たさなければならない。もう1つが、2017年に導入された「医薬品条件付き早期承認制度」である。これは、患者数が少なく、治療の選択肢が限られ、かつ検証臨床試験の実施が難しいため医療上の大きなニーズが満たされない疾病への治療薬を対象とするものである。この制度により、製薬企業は、製販後に有効性・安全性の再確認等のために必要な調査等を実施すること等を条件として、検証的臨床試験以外の臨床試験などによって一定程度の有効性及び安全性が示されたデータを元に承認申請を行えるようになった。既にいくつかの医薬品が本制度の対象品目となっている[15]。
医療用医薬品の安全性と品質の担保
他の主要国同様に、日本における医薬品の開発・製造・販売においては、GLP、GCP、製造所における製造管理・品質管理の基準(GMP: Good Manufacturing Practice)、製造販売後安全管理の基準(GVP: Good Vigilance Practice)等を含む医薬品の開発から工場管理等までの薬事規制に関する各基準(GXP: Good X practice)によって統制されている。他方で、製品の品質及び安全性に関して、日本の規制は、いくつかの独自の特徴を有している。
第一に、薬機法の下で日本の医薬品・医薬部外品、化粧品及び再生医療など製品の品質管理の基準(GQP: Good Quality Practice)とGVP要件を満たすために、製薬会社は製品の品質と安全性にかかわる活動を監督し統制する3名の責任者(通称:三役)を任命しなければならない[16]。
- General marketing compliance officer (総括製造販売責任者):品質管理及び製造販売後安全管理のために必要な業務を遂行し、全体責任を負う者。
- Quality assurance manager (品質保証責任者):GQPの規則に従って、品質管理業務を適正かつ円滑に遂行しうる能力を有する者。
- Safety manager (安全管理責任者):GVPの規則に従って、安全確保業務を適正かつ円滑に遂行しうる能力を有する者。
第二に、新薬承認後には「市販直後調査」が義務付けられている[17]。すなわち、新薬が承認された後、厚労省は製薬会社に、患者の安全性を追跡し確保するために医薬品の製造販売後の調査活動を行うことを義務付けており、調査活動は通常、新薬の上市後6ヵ月間実施する。この調査は、日本の医薬品製造販売後調査・試験の実施の基準(GPSP: Good Post-marketing Surveillance Practice)に準拠して、臨床試験中には観察されなかった可能性のある重篤な副作用が現実の使用において起きていないかを追跡することを目的とする[18]。
第三に、さらに患者の安全を確実に確保するため、新薬は原則として、収載された月の初日から1年間は処方日数が14日までに制限される。一方、以下2つの基準のいずれかを満たす医療用医薬品の場合は、14日の処方日数上限を適用しない特例が中医協によって認められている[19]。
- 1年以上の臨床使用経験がある既収載品と実質的に同様の効能・効果、用量・用法の有効成分を配合した新薬(例えば新医療用配合剤等)
- 疾病の特性や製剤上の特性により、含有量が14日を超える製剤のみが存在しており、投薬期間が14日を超えることの安全性が確認されている新薬
先発医薬品の薬価決定[20]
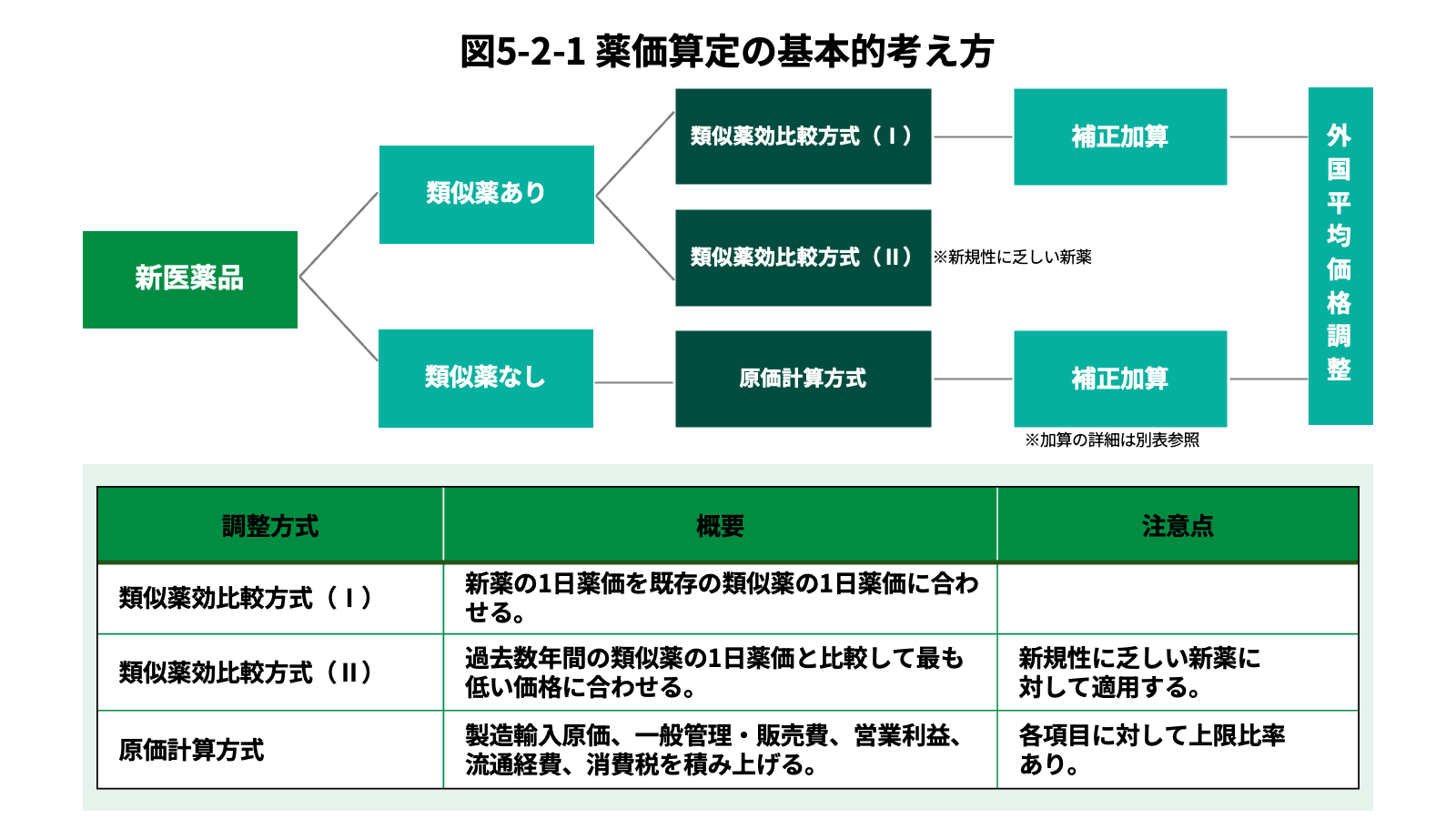
薬価とは、保険医療機関及び保険薬局が、薬剤の支給に要する単位あたりの平均的な費用の額として銘柄毎に定める額をいう。はじめに、収載時の薬価の決定プロセスについて説明する。市場に既に承認された類似薬が存在する場合は「類似薬効比較方式」で、承認された類似薬が存在しない新薬の場合は、「原価計算方式」で決定する[21]。更に、革新性が限定的と評価された医薬品の薬価を抑制しつつ、革新性の高い医薬品開発を奨励する目的で、以下表の通り、薬価への補正加算制度も導入されている[22][23]。また、公正な市場競争を確保する観点から、原価計算方式又は薬理作用類似薬のない品目における類似薬効比較方式において、諸外国における価格との乖離が大きい場合(外国平均価格の1.25倍以上又は0.75倍以下)に、価格の調整を行う「外国平均価格調整」も設けられている[24]。
| 補正加算の種類 | 概要 | 加算率 |
|---|---|---|
| 画期性加算 | 新規の作用機序、高い有効性・安全性、疾病の治療方法の改善 | 70~120% |
| 有用性加算 | 高い有効性・安全性、疾病の治療方法の改善 等 | 5~60% |
| 市場性加算 | 希少疾病用医薬品 等 | 5~20% |
| 小児加算 | 用法・用量に小児に係るものが明示的に含まれている 等 | 5~20% |
| 特定用途加算 | 特定の用途で高い有用性が認められる | 5~20% |
| 先駆加算 | 先駆的な用途で高い有用性が認められる | 10~20% |
| 迅速導入加算[25] | 革新的新薬を日本へ迅速に導入した | 5~10% |
先発医薬品の薬価改定
次に、薬価収載された後の改定について概説する。薬価改定は、市場価格と薬価との乖離率等を理由に1年に一度実施される[26]。具体的には、保険医療機関・保険薬局や医薬品卸売販売業者を対象とした医薬品価格調査で得られた納入価格の加重平均値に流通安定のための調整幅と消費税を加味することにより、新薬価が算定される。
薬価と実勢価格の乖離は医療機関や薬局の収益となっていることを踏まえ、薬価は、各品目の市場実勢価格の加重平均値に調整幅(2%)を加えた額に改定することにしている[27]。言い換えれば、薬価と市場実勢価格の加重平均値に2%以上の差が生じた場合、その差分だけ薬価を改定する仕組みである。この薬価改定には、原則的には医薬品の価格を引き下げる効果があり、厚労省はこの制度によって全体的な医療費支出を抑制していると言える。
その他、市場が大幅に拡大したり特許切れとなったりした医薬品の価格を下げることで、医療費全体の増加を抑制している。さらには新薬の開発を奨励するため、医薬品の利益率の確保を目的とした追加的に再算定を行う制度[28]も設けている(詳細は下表を参照)。その他にも、費用と効果のバランスの観点から価格の一部を調整する費用対効果評価制度も導入されている。医療費が右肩上がりで増える中、薬価を取り巻く制度は今後も更なる変更が予想される。
| 再算定 | 種別 | 説明 |
|---|---|---|
| 長期収載品への追加引き下げ | 引き下げ | 長期収載品(特許切れ新薬)の追加引き下げを行う。新薬開発を促す薬価引き上げの財源とする |
| 市場拡大再算定 | 引き下げ |
年間販売額が予想販売額よりも大きく拡大した場合、一定の条件の下、薬価を引き下げる。 ※中医協であらかじめ特定した領域に該当する品目は、再算定を行わない。 |
| 効能変化再算定 | 引き下げ | 主たる効能・効果の変更がなされた医薬品であって、変更後の主たる効能・効果に係る類似薬があるものについて、変更後の効能・効果の類似薬の価格に近づくよう、薬価を再算定を行う。 |
| 用法用量変化再算定 | 薬品による | 主たる効能・効果に係る用法・用量に変更があった品目については、変更前後で、1日薬価が同額となるよう再算定を行う。 |
| 不採算品再算定 | 引き上げ |
保険医療上の必要性が高いものであると認められる医薬品であって、薬価が著しく低額であるため製造販売業者が製造販売を継続することが困難であるもの等については、原価計算方式によって算定される額に改定する。 ※その際、営業利益率は100分の5を上限とする。 |
| 新薬創出・適応外薬解消等促進加算 | 引き上げ | 革新的新薬の創出、ドラッグ・ラグ/ロスの解消を促進するため、新薬の市場実勢価格に基づく薬価の引下げを猶予する。適用にあたっては、企業要件と品目要件を満たす必要がある。 |
- [12] 独立行政法人医薬品医療機器総合機構「新医薬品(医療用医薬品)」https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/p-drugs/0021.html
- [13] 独立行政法人医薬品医療機器総合機構「新医薬品に係る承認審査の標準的プロセスにおけるタイムライン)」https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/p-drugs/0014.html参照。なお、当該ページより、承認審査プロセスの各工程における所要期間の目安を確認することも可能である。
- [14] 厚生労働省「先駆的医薬品指定制度について」https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iyakuhin/tp150514-01_00001.html
- [15] 厚生労働省「医薬品の条件付き承認制度について」 https://www.mhlw.go.jp/content/10601000/000954228.pdf
- [16] 厚生労働省「医薬品の製造販売業者における三役の適切な業務実施に関する留意事項(別添)」https://www.mhlw.go.jp/content/11120000/000804704.pdf
- [17] 市販直後調査制度の詳細については、厚生労働省「医療用医薬品の市販直後調査の実施方法等について」https://www.mhlw.go.jp/content/11120000/000945083.pdf 参照。
- [18] 独立行政法人医薬品医療機器総合機構「市販直後調査に関する情報」https://www.pmda.go.jp/review-services/drug-reviews/review-information/p-drugs/0006.html
- [19] 厚生労働省「新医薬品の処方日数制限の取扱いについて 」https://www.mhlw.go.jp/content/12404000/001242877.pdf(p.4)。
- [20] 詳細は、毎年2月に発出される厚生労働省保健局通知「薬価算定の基準について」を参照。執筆時点での最新版は、令和7年2月19日発出「薬価算定の基準について」https://www.mhlw.go.jp/content/12404000/001416107.pdfである。
- [21] 厚生労働省「現行の薬価基準制度(概要)」https://www.mhlw.go.jp/content/10808000/001276014.pdf
- [22] 厚生労働省「現行の薬価基準制度(概要)」https://www.mhlw.go.jp/content/10808000/001276014.pdf
- [23] この際、比較方式と原価計算方式のいずれの方式で薬価を算定する場合にも、補正加算の対象となる。
- [24] 厚生労働省「現行の薬価基準制度(概要)」https://www.mhlw.go.jp/content/10808000/001276014.pdf
- [25] 2024年度に新設された加算。詳細は、厚生労働省「現行の薬価基準制度(概要)」参照。
- [26] 厳密には、奇数年の薬価改定(いわゆる中間年改定)は2021年度から始まったが、偶数年の本改定と比較して適用される薬価算定ルールは限定的であった。しかし、奇数年度の改定についても偶数年における本改定時での薬価算定ルールに近づきつつある。(参照:厚生労働省「令和7年度薬価改定について(参考資料)」https://www.mhlw.go.jp/content/10808000/001360022.pdf)以上を踏まえて、本稿では本改定の基準について概説する。
- [27] 厚生労働省「現行の薬価基準制度(概要)」https://www.mhlw.go.jp/content/10808000/001276014.pdf
- [28] 厚生労働省「現行の薬価基準制度(概要)」https://www.mhlw.go.jp/content/10808000/001276014.pdf
5.3 後発医薬品
後発医薬品の定義
先発医薬品の特許が切れた後に、先発医薬品と成分や規格が同一で、治療学的に同等であるとして承認される医薬品を後発医薬品(ジェネリック医薬品)と呼ぶ。先発医薬品には特許制度によって独占的に製造・販売できる期間(特許の有効期間及び有効性・安全性を検証する再審査期間)が定められているが、その期間以降に後発医薬品の申請・販売が可能となる。
後発医薬品は、先発医薬品と効能・効果、用法・用量が原則的に同一である一方、先発医薬品に比べて低価格であるという特徴がある。また、先発医薬品が最初に開発したメーカーの独占であるのに対して、後発医薬品は複数のメーカーが製造・販売するケースも多い[29]。
後発医薬品の承認
後発医薬品を製造・販売するにあたっては、先発医薬品の場合と異なり、有効成分の有効性・安全性を検証するための治験を行う必要はない。ただし、同じ有効成分をもつ薬であっても、製造工程などが異なれば同じように作用しない可能性があることから、後発医薬品の製造・販売においては先発医薬品と有効性・安全性が同等であるかどうかについて審査される。審査内容は、主に「規格及び試験方法」「安定性試験」「生物学的同等性試験」の3項目であり、これらの審査によって後発医薬品が先発医薬品と同等であることが示されれば、有効性・安全性に問題がないことが確認され、承認されることとなる。
医療用後発医薬品においては、製造販売承認申請資料に係る不正事案や、製造・品質管理体制の不備に伴う品質問題が発生し、社会の注目を集めている。2021年以降、承認申請者の責任をより明確にし、承認申請資料の信頼性を確保するため、後発医薬品の承認審査において、製造者は承認申請時の添付資料として新たに承認申請資料の信頼性に係る説明資料の提出が必要になると共に、製造所の人員配置の状況と妥当性について確認を受けるといった新たなルールが設けられている[30]ため、注意が必要である。
後発医薬品の薬価[31]
後発医薬品は治験が不要であることから、研究開発費用が先発医薬品に比べて大幅に少なくて済むため、製薬会社としては先発医薬品よりも安い価格設定であっても、利益を確保することが可能である。そのため、日本の公的医療保険制度においては、後発医薬品の償還価格は、先発医薬品の50%以下という大幅に安い水準に設定されることが一般的である。
また日本の公的医療保険制度においては、薬価を決定する際に、市場の実勢価格を参照する。そのため、同じ有効成分を含む後発医薬品が複数あり、市場での価格競争が起きれば、それを反映してより安価に設定されることとなる。
後発医薬品が承認され、公的医療保険の適用対象として薬価基準に収載(保険適用)されるタイミングは、年に2回(6月と12月)ある。後発医薬品が開発され、初めて保険適用となる場合、基本的には先発医薬品の薬価の50%となる。ただし例外規定が2つある。第一に、内服薬については、薬価基準への収載を希望する同種の製品が同じタイミングで10品目を超えた場合、先発医薬品の薬価の40%となる。これは、1つの先発品に対して多くの後発品が販売されるのを避ける意図がある。第二に、バイオ後続品(バイオシミラー)については先発医薬品の70%となる。なお、後発医薬品が既に収載されている場合、最低価格の後発品と同価格となる。
さらにそもそも薬価制度では、最初に薬価基準に収載された後、市場実勢価格の変化に基づき、毎年実施される薬価改定において、少しずつ引き下げられることとなっている。そのため、複数の後発医薬品があるケースにおいては、以下の通り価格帯ごとに一つの価格を設定する措置がとられている。
- 最高価格の30%を下回る算定額となる後発品を一つの価格(加重平均)として収載
- 最高価格の30%以上、50%を下回る算定額となる後発品を一つの価格(加重平均)として収載
- 最高価格の50%以上の算定額となる後発品を一つの価格(加重平均)として収載
近年は、収載から12年経過した後発品は、原則として、加重平均により1つの価格帯に集約する措置や、価格帯の集約により改定前より薬価が引き上がることを抑制するための措置を導入し、薬価のばらつきを防ぎ、引き下げが進む仕組みになっている。
後発医薬品の処方・調剤
既に述べたように、厚生労働省は後発医薬品の使用を政策的に推進している。日本の公的医療保険制度では、医師が医薬品を処方する際には、有効成分の一般名を用いることが原則である。薬局は、医師からの特段の指示がない限り、患者への説明と同意を得たうえで後発医薬品を使用することが推奨されており、後発医薬品を積極的に調剤する薬局に対するインセンティブも設定されている。もっとも、医師が処方箋に署名等を行えば、先発医薬品を指定して後発医薬品への変更をしないように指示することもできる。
なお、後発医薬品の処方・調剤について、日本と同様の仕組みを採っている国としては、フランスが挙げられる。フランスでは、薬局における後発医薬品による代替調剤を拒否した場合には、患者が薬局で全額自己負担をしたうえで、別途償還の申請が必要となる仕組みを導入するなど、後発医薬品の使用を強力に推進している。
一方、日本と異なる仕組みを採っている国としては、アメリカが挙げられる。アメリカでは、患者が契約する医療保険の内容によって使用できる医薬品が決まっているため医師や薬局による自由度は低く、どの医薬品を採用するかについては、保険者であるHMO(Health Maintenance Organization)と製薬会社との交渉によって決定している。
後発医薬品の産業政策
日本政府は、国民皆保険制度を維持し、医療の質を確保しながら効率的な医療サービスの提供を継続するため[32]、後発医薬品の使用を促進する方針を採っている。具体的には、2021年6月の閣議決定(「経済財政運営と改革の基本方針 2021」)において、後発医薬品の数量シェアを、2023年度末までに全ての都道府県で80%以上とするという目標を定めていた。その結果、2023年3月時点で、36都道府県がこの目標を達成している。また、新たに後発医薬品の金額シェアを用いた目標として、「2029 年度末までに、後発医薬品の薬価総額を後発医薬品の薬価総額及び後発医薬品のある先発医薬品の薬価総額の和で除した割合を 65%とする」ことを掲げている[33]。今後も後発医薬品の利用促進が進められていくと考えられる。
- [29] このような事情を背景に、欧米では、商品名で呼ばれることの多い先発医薬品と対比して、有効成分の一般名(generic name)で処方されることが多いことから、「generic drug」という言葉で呼ばれており、日本でも「ジェネリック医薬品」と呼ばれるようになっている。
- [30] 厚生労働省「医療用後発医薬品の承認審査時における新たな対応について」https://www.pmda.go.jp/files/000250546.pdf
- [31] 本項目は、厚生労働省「令和6年度薬価改定について 4~ 後発品・長期収載品〜」https://www.mhlw.go.jp/content/12404000/001129375.pdfのうち、後発医薬品の価格改定に係る概略を整理したものである。
- [32] 背景として、日本では国民皆保険制度により、一定の自己負担で必要な良質の医療を平等に受けることができる一方で、医療技術の進歩、急速な高齢化等の要因により医療費は増加し続け、医療財源が逼迫しつつあることから、国民皆保険制度の継続が不安視されていることがある。
- [33] 厚生労働省「安定供給の確保を基本として、後発医薬品を適切に使用していくためのロードマップ」https://www.mhlw.go.jp/bunya/iryou/kouhatu-iyaku/dl/roadmap04.pdf
5.4 OTC医薬品
医療用医薬品とOTC医薬品のすみわけ
医薬品には、Section 5.2及び5.3で詳述した医療用医薬品以外に、一般の人が直接薬局や薬店等で購入し、自らの判断で使用できる一般用医薬品と要指導医薬品が存在する。なお、一般用医薬品と要指導医薬品を合わせて、「OTC医薬品」と呼ぶ。この名称は、薬剤師が薬局の店頭にてカウンター越しに販売する様子から、「Over The Counter:オーバー・ザ・カウンター」に由来しているとされる。
一般用医薬品とは、医療用医薬品として取扱われる医薬品以外の医薬品(要指導医薬品を除く)をいう。すなわち、一般の人が薬局等で購入し、自らの判断で使用する医薬品であって、通常、安全性が確保できる成分の配合によるものが多い[34]。近年日本では、一般用医薬品についてはインターネット販売が可能となった。また、一般の消費者が自らの判断で購入・服薬できるため、販売者に対して購入者への情報提供が義務付けられており、そのリスク区分により、第1類、第2類、第3類の3種に分けられ、情報提供の必要性の程度が定義されている。
- 第1類医薬品:一般用医薬品としての安全性評価が確立されておらずリスクが不明な医薬品、あるいは日常に支障を来す副作用のおそれがあり、特に注意が必要なもの。
- 薬剤師による情報提供が義務づけられており、薬剤師の管理・指導の下でのみ販売や受け渡しが可能である。
- 第2類医薬品:日常生活に支障を来す副作用のおそれがある医薬品。
- 薬剤師または登録販売者の管理・指導の下でのみ販売や受け渡しが可能で、かつ情報提供は努力義務とされるもの。
- 第3類医薬品:その他の医薬品
- リスクの程度は一番低く、購入者から直接希望がない限り、販売に際しての商品説明について法的制限をうけないもの。
要指導医薬品は、2013年の薬機法改正で新たに設けられた分類であり、患者の選択によって使用できる一方、薬剤師の対面での指導・情報提供を必要とする医薬品を指す[35]。例えば医療用から一般用に移行して間もなく、一般用医薬品に分類できるだけのリスク情報が明らかになっていない医薬品(いわゆる「スイッチ直後品目」)や、医療用医薬品を経ずに直接OTC医薬品として承認された一般用医薬品、および劇薬等が該当する[36]。
| OTC医薬品分類 | 対応する専門家 | 販売者からお客様への説明 | お客様からの相談への対応 | インターネット、郵便等での販売 | |
|---|---|---|---|---|---|
| 要指導医薬品 | 薬剤師 | 対面で書面での情報提供(義務) | 義務 | 不可 | |
| 一般用医薬品 | 第1類医薬品 | 書面での情報提供(義務) | 可 | ||
| 第2類医薬品 | 薬剤師または登録販売者 | 努力義務 | |||
| 第3類医薬品 | 法律上の規定なし | ||||
- [34] 厚生労働省「医療用医薬品と一般用医薬品の比較について」https://www.mhlw.go.jp/shingi/2004/09/s0906-6c.html
- [35] 厚生労働省「要指導医薬品について」https://www.mhlw.go.jp/content/11121000/001062522.pdf による定義では、「その効能及び効果において人体に対する作用が著しくないものであって、薬剤師その他の医薬関係者から提供された情報に基づく患者の選択により使用されることが目的とされているものであり、かつ、その適正な使用のために薬剤師の対面による情報の提供及び薬学的知見に基づく指導が行われることが必要なものを指す。」とされている。
- [36] 厚生労働省「要指導医薬品について」https://www.mhlw.go.jp/content/11121000/001062522.pdfでは、以下の4つの類型が示されている。(1)新医薬品であって、再審査期間中のもの (医療用医薬品を経ずに直接OTCとして承認された品目) (2)医療用医薬品から転用された医薬品であって、製造販売後調査期間中のもの (スイッチ直後品目)(3)薬機法第44条第1項に定める毒薬及び薬機法第44条第2項に定める劇薬。
5.5 医療機器
医療機器の分類と承認[37]
医療機器とは、構造、使用方法、効果又は性能が明確に示されるものであって、「疾病の診断、治療、予防に使用されること」又は「身体の構造、機能に影響を及ぼすこと」のどちらかの目的に該当し、政令で定めるものである[38]。医療機器は使用における安全上のリスクや目的や用途などの種別により、以下の4つの管理上のクラスに分類されており、クラスごとでその承認条件が異なる。特に人体へのリスクの高いクラスIII及びクラスIVはPMDAの審査が必要になる。
| クラス分類 | 該当する医療機器 | 規制 | |
|---|---|---|---|
| 一般医療機器 | クラスI |
|
|
| 管理医療機器 | クラスII |
|
|
| 高度管理医療機器 | クラスIII |
|
|
| クラスIV |
|
|
|
医療機器承認審査の迅速化
近時、医療機器の承認審査等の迅速化のため、厚労省と製造販売業者双方の協力により、様々な取り組みを行う共同計画を策定・実施している。具体的な制度を紹介する。
2019年には「医療機器規制と審査の最適化のための協働計画」と「体外診断用医薬品規制と審査の最適化のための共同計画」が策定され、医療機器等の開発プロセスと規制の最適化を目指し、企業が承認申請するまでの開発ラグの解消や各種規制の国際整合をさらに進めることを目標とした。更に、2019年の薬機法改正では、AIを活用した医療機器など市販後に性能が変化することが前提とされる医療機器について、事前に計画された範囲の中で承認事項の一部変更が認められる迅速な承認審査制度が創設された。また、2021年には「プログラム医療機器実用化促進パッケージ戦略」(DASH for SaMD)に基づき、厚労省とPMDAに窓口を設置して対応を強化した。2023年には、「プログラム医療機器実用化促進パッケージ戦略2」(DASH for SaMD2)を公表し、イノベーションの早期把握と審査の考え方の公表、二段階承認の考え方の公表等をはじめとしたプログラム医療機器の特性を踏まえた実用化促進に向けた取り組み、PMDAの相談体制の強化・海外展開支援等を行なっている。
医療機器の保険償還[40]
医療機器は医療用医薬品とは異なり、承認された医療機器がすべて保険適用となるわけでは無い。保険適用対象となる医療機器は保険医療材料として指定される必要がある。保険医療材料には、診療報酬において償還することのできる特定治療材料[41]や、技術料の加算または技術料に包括して評価されている治療材料[42]が含まれる。
保険医療材料である場合、以下に示す評価区分のいずれに属するかによって、その価格の設定及び保険請求における取扱いが異なる。評価区分の決定は、薬事承認後に「保険適用希望書」を提出し、必要に応じて保険医療材料等専門組織による検討及び中医協による承認を経て行われる。決定された評価区分に沿って保険が適用される。
- 「A1(包括)」「A2(特定包括)」及び「A3(既存技術・変更あり)」に該当する保険医療材料は、診療報酬項目(技術料)の中に当該製品の価格も含まれており、診療報酬項目と別に製品の価格を保険請求することができない。
- 「B1(既存機能区分)」「B2(既存機能区分・変更あり)」及び「B3(期限付改良加算)」に該当する保険医療材料は「特定保険医療材料」と呼ばれており、機能区分ごとに製品の価格(基準材料価格)が決められており、技術料(診療報酬項目)とは別に保険請求することができる。
- 「C1(新機能)」及び「C2(新機能・新技術)」に該当する医療保険材料は、B区分と同様、技術料(診療報酬項目)とは別に製品の価格を保険請求することができる。しかし、承認時に、既存の機能区分に該当しないため、新たな機能区分が必要であり、また、「C2(新機能・新技術)」においては、それを用いる技術も診療報酬項目に収載する必要もある。
- B3、C1、及びC2区分に該当する医療機器は中医協における了承が必要である。
| 保険医療材料の評価区分 | 評価方法 | |
|---|---|---|
| A1 | 包括 | 既存の診療報酬項目において包括的に評価 |
| A2 | 特定包括 | 既存の特定の診療報酬項目において包括的に評価 |
| A3 | 既存技術・変更あり | 当該製品を使用する技術を既存の診療報酬項目において評価(留意事項等の変更を伴う) |
| B1 | 既存機能区分 | 既存の機能区分により評価され、技術料とは別に評価 |
| B2 | 既存機能区分・変更あり | 既存の機能区分により評価され、技術料とは別に評価(機能区分の定義等の変更を伴う) |
| B3 | 期限付改良加算 | 既存の機能区分に対して期限付改良加算を付すことにより評価 |
| C1 | 新機能 | 新たな機能区分が必要で、それを用いる技術は既に評価 |
| C2 | 新機能・新技術 | 当該製品を使用する技術が未評価 |
| R | 再製造 | 再製造品について、新たな機能区分により評価 |
| F | 保険適用に馴染まないもの | ー |
新規医療材料の基準材料価格[43]
特定保険医療材料は機能区分ごとに保険償還価格が定められており、これを「機能区分別収載」と呼ぶ[44]。機能区分別収載においては、各機能区分内の製品の保険償還価格は全て同一価格で設定されている。
新規医療材料の基準材料価格は、類似機能区分があれば「類似機能区分比較方式」、類似機能区分がなければ「原価計算方式」で設定されており、診療報酬・薬価と同じタイミングで改定される[45]。また、薬価同様に、革新性が限定的と評価された医療材料の価格を抑えつつ、革新性の高い技術開発を奨励する目的で、補正加算制度も導入されている。更に、公正な市場競争を確保する観点から、外国平均価格調整[46]も行われている。
基準材料価格の改定においては、医薬品と同様、実際の市場価格との乖離率を基に改定をされている。基本的には、市場実勢価格に消費税を加えた算定値に一定幅(4%)を加算した額とし、改定前の基準材料価格を超えないこととしている[47]。
また、厚労省は基準材料価格において、イノベーションを評価する制度も導入している。
- チャレンジ申請:使用実績を踏まえて保険適用後に新規機能区分の該当性について再度評価を行うことができる仕組み
- 期限付改良加算:既存機能区分の既収載品と置き換わり得る製品に対して、同一機能区分としつつ、当該製品が新規収載されてから2回の改定を経るまで時限的に加算する仕組み
- 機能区分の特例:革新性の高い製品や先駆け審査指定を受けた製品について、保険適用されてから2回の改定を経るまで、他の既収載品とは別に材料価格改定等を行い、後から申請するB区分製品の価格の影響を抑制する仕組み
また、医療機器においても、2019年以降、医療用医薬品と同様により適正な償還価格を設定できるよう、償還価格の決定における費用対効果に基づく価格調整方法が導入されている。なお、費用対効果評価制度の対象は市場規模が大きい、又は著しく単価が高い医薬品・医療機器のみであり、また評価結果は保険償還の可否の判断に用いるのではなく、いったん保険適用したうえで価格調整に用いるとされている[48]。
現在、「迅速な保険適用の運用を維持した上で、イノベーションの推進や現役世代等の保険料負担に配慮する観点から、費用対効果評価のさらなる活用の在り方について、医薬品の革新性の適切な評価も含め、検討する」こととする政府方針に基づき、費用対効果評価制度の更なる活用について、厚生労働省の審議会にて協議が進んでいる[49]。
医療機器産業政策
医療機器産業は、先進国における今後の主力産業の一つとして期待されており、日本でも同様である。日本は、内視鏡や画像診断機器等、一定の世界シェアを有する領域がある一方で、後塵を拝している分野も見られる。そこで、政府は2016年の閣議決定にて、医療機器の研究開発と普及に関して実施すべき基本計画を取りまとめた[50]。2022年には、新型コロナ禍で顕在化した安定供給の確保等を盛り込んで、2期目の基本計画が閣議決定された[51]。この閣議決定に基づき、様々な事業等によって医療機器産業に対する支援が行われている。
- [37] 独立行政法人医薬品医療機器総合機構「医療機器」https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/devices/0028.html
- [38] 薬機法第2条第4項にて、「この法律で「医療機器」とは、人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は人若しくは動物の身体の構造若しくは機能に影響を及ぼすことが目的とされている機械器具等(再生医療等製品を除く。)であつて、政令で定めるものをいう。」と定められている。
- [39] 2014年11月25日より、クラスIIIの高度管理医療機器のうち、認証基準のあるものについては、第三者認証機関による認証が可能となっている。
- [40] 厚生労働省「令和6年度保険医療材用制度改革の骨子 参考資料」https://www.mhlw.go.jp/content/12404000/001180647.pdf
- [41] 特定治療材料とは、中医協「特定保険医療材料の評価に関する建議書」https://www.ipss.go.jp/publication/j/shiryou/no.13/data/shiryou/iryou/603.pdfによると、(1)医科診療報酬関係(老人診療報酬を含む。)、(2)歯科診療報酬関係(老人診療報酬を含む。)、(3)調剤報酬関係(老人診療報酬を含む。)を指す。
- [42] PMDA「医療機器の保険適用について」 https://www.pmda.go.jp/files/000273983.pdf
- [43] 厚生労働省「保険医療材料制度の概要」https://www.mhlw.go.jp/content/10808000/001508753.pdf
- [44] Section 5.2、5.3で概説したように、医療用医薬品は銘柄ごとに保険償還価格が定められているため、保険償還価格決定の枠組みが異なることに注意が必要である。
- [45] 厚生労働省「平成 28 年度保険医療材料制度改革の概要」http://www.mhlw.go.jp/file/06-Seisakujouhou-12400000-Hokenkyoku/0000114377.pdf
- [46] 2024年度の保険医療材料制度改革以降、保険償還価格が外国平均価格を一定程度上回る場合に再算定の対象となる方式を用いて、再算定に係る外国価格調整の比較水準等を行っている。参照 厚生労働省「令和6年度保険医療材料制度改革の概要」https://www.mhlw.go.jp/content/12400000/001251540.pdf
- [47] 厚生労働省「令和6年度保険医療材料制度改革の概要」https://www.mhlw.go.jp/content/12400000/001251540.pdf
- [48] 厚生労働省「令和6年度診療報酬改定の概要【費用対効果評価制度】」https://www.mhlw.go.jp/content/12400000/001251541.pdf
- [49] 厚生労働省「中央社会保険医療協議会費用対効果評価専門部会」https://www.mhlw.go.jp/stf/shingi/shingi-chuo_128159.html
- [50] 閣議決定の概要につき、首相官邸「国民が受ける医療の質の向上のための医療機器の研究開発及び普及の促進に関する基本計画(概要)」https://www.kantei.go.jp/jp/singi/kenkouiryou/kaihatsu/dai4/sankou1.pdf
- [51] 閣議決定の概要及び本文につき、厚生労働省「「国民が受ける医療の質の向上のための医療機器の研究開発及び普及の促進に関する基本計画」の変更について」https://www.mhlw.go.jp/stf/newpage_25953.html